Keywords: nitrone nitroxide cerovive renovis astrazenica stroke ischaemic injury pbn
Incorporated by reference in US patent 5,723,502, covering therapeutic use of spin traps. Note "stroke" in table 1
RedoxSignaling.com.. Nitrone.com.. Earlier review.. Later Review
Physiological Chemistry and Physics and Medical NMR, 16 (1984),175-195
A Review
FREE RADICALS AND DISEASE IN MAN
PETER H. PROCTOR, PhD. MD* and EDWARD S. REYNOLDS, MD**,
Abstract:
Free radicals and related activated electronic species are produced in biological systems in antimicrobial defense, through the action of the mixed function monooxygenases, by various oxidative enzymes such as xanthine oxidase, and by autooxidations mediated by such agents as heavy metals or quinones. While the evidence is circumstantial, £xcessive unconfined or inappropriate production of radical species in inflammation, the metabolism of exogenous chemicals, or through autooxidation probably plays a significant role in human disease.
Introduction.
Free radicals are short-lived reactive chemical species having one or more electrons with unpaired spins. Previous authors (1-44) have extensively reviewed the evidence for roles for such electronicallyactivated species (e.g., superoxide and singlet oxygen) in the normal function of cells and tissues and in the etiology of certain diseases in man. Radical generating processes may be key components in the toxicity of many drugs, (8,9,11,20,22-28.34,42,47-52) in antimicrobial defense, (5,6,10,34,38-46) and in inflammation.(7,32,41) This review, restricted to "acute" pathologic processes such as inflammation and drug toxicity, broadly summarizes our current understanding of this field, its seductive and circumstantial nature, and its clinical relevance. Since an in-depth analysis of any of the individual areas of this field could amount to a book in itself, we have made certain other arbitrary limitations on this review.
For example, for the roles of free radicals in aging and in carcinogenesis, the concerned reader is referred to other authors, while Table I lists a few events relevant to the role of active mechanisms in human disease. Likewise, to cover such broad areas comprehensibly in a brief review requires that the reader refer to previous authors (l-44) for more detailed analysis of each individual area. Hopefully, the resulting broad synthesis justifies such liberties.
The Nature of Radical and Excited-state Species. (See Glossary).
Moving charges generate magnetic fields. An orbital electron may be viewed as a moving negative charge which generates a magnetic field ("magnetic moment''). Depending on the direction of its motion ("spin") about the orbital axis, this magnetic field will have its north magnetic pole oriented either "up" or "down" relative to its orbit. Logically, this magnetic property of the electron is called "spin" (Figure 1).
Stable compounds, especially those composed of low atomic weight atoms, have even numbers of electrons arranged two to each "orbital". The two electrons in each orbital are "paired". That is, their "spin"-derived magnetic fields are of opposite polarity, effectively canceling them.
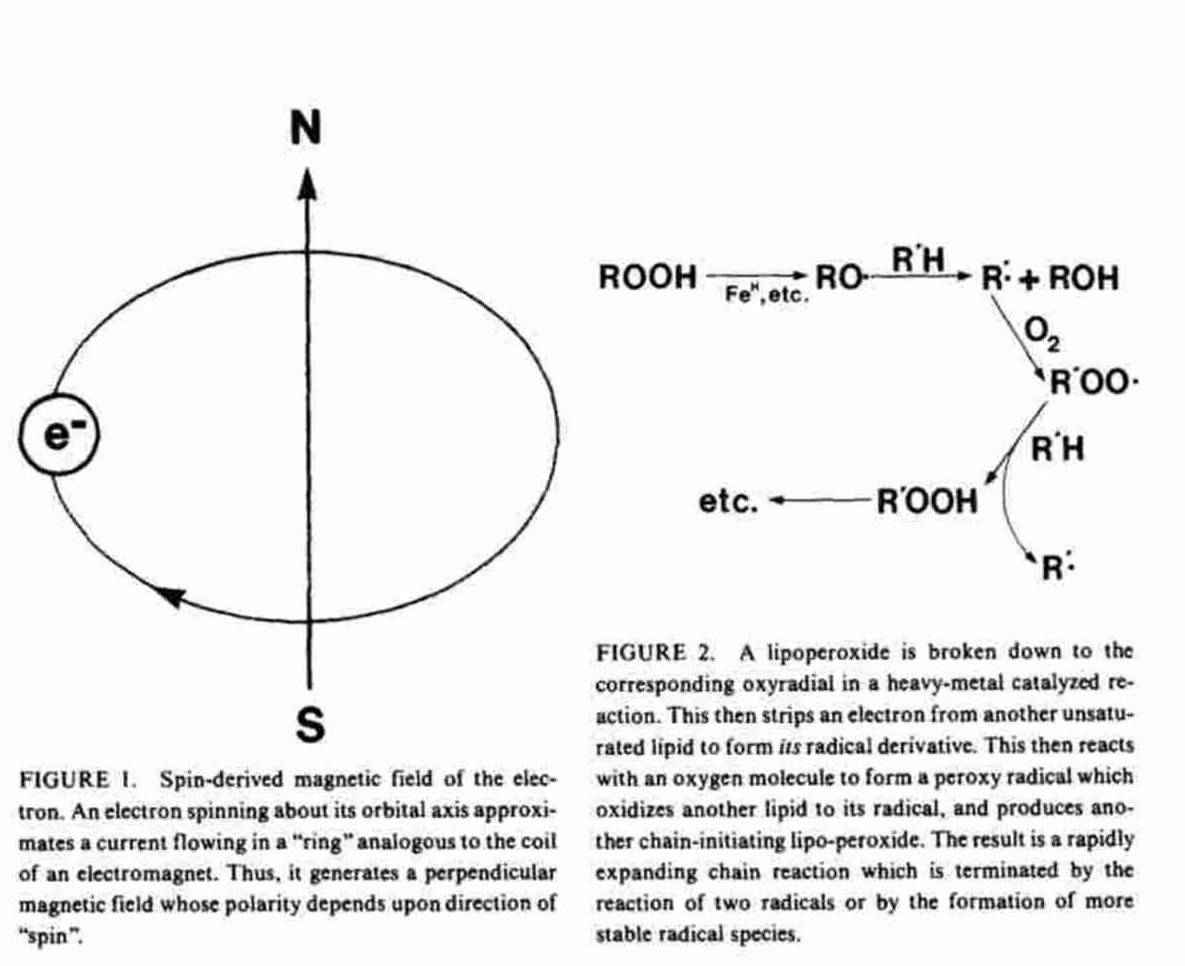
A free radical with one or more of its electrons unpaired has either an odd number of orbital electrons, with one unpaired - a free radical per se - or of pairs of electrons of the same spin isolated singly in separate orbitals. Molecular oxygen is an example of such a biradical. (16,17) These uncancelled spins give radical species a net magnetic moment which can be directly detected by electron spin resonance (ESR) spectroscopy, a technique which involves aligning the unpaired electron with an external magnetic field and then measuring its relaxation as the field is removed.(23,27,28), A compound may become a free radical either by gaining (e.g., superoxide radical) or by losing (e.g., ascorbate radical) a single electron in a reduction/ oxidation reaction. Such a reaction produces the radical products of both molecules, and can result in radical chain reactions.(16-19) The rate of propagation of radical chain reactions depends upon the reactivity of the radicals formed with the substrates available.(15-21), Figure 2 shows an example of such a chain reaction, the oxidative degradation of unsaturated fatty acids.
In biological systems radical species tend to be restricted to derivatives of molecular oxygen (Figure 3), polyunsaturated fatty acids (Figure 2), sulfhydryl compounds, quinones or quinone-like compounds including flavins (Figure 5) and other compounds which can easily transfer single electrons.
Free radicals are generally highly reactive and participate in hydrogen abstraction, radical addition, bond scission, and annihilation reactions. For example, they can oxidize unsaturated fatty acids in cell membranes,(15-21( damage DNA, (53-57) oxidize protein amino acid side-chains,(16-19,58,59) depolymerize hyaluronic acid,(60) modulate nucleotide cyclase activities,(61-67) and the action and synthesis of prostaglandins and lipoperoxides.(48,68-71). In short, radical species can attack most biological substrates from large macromolecules to smaller molecules such as catechols. Exceptionally, stable radical species can be formed, mostly due to steric protection of the unpaired electron. Examples include the melanins (72-75)and the stable radical derivatives produced in spin-trapping measurements.(7,28)
TABLE I. Historical perspective of biological free radicals (a few representative events)
1800's -- Vitreous chalchosis(22O) association between deafness and pigmentary abnormalities(243)
1920's -- Radical mechanisms responsible for radiation and oxygen toxicity
1938 -- Discovery of superoxide(259)
1938 -- Respiratory burst of granulocytes(103)
1939 -- Isolation and characterization of cupreins(260)
1950 -- Protein antiinflammatory contaminant of hyaluronidase (Wydase) recognized by Schulte(l84)
1952 -- Application of ES R to biological materials(261)
1958-64 -- Free radical metabolites of the phenothiazines,(77,80) the melanins(73-75), Hormones(88), carbon tetrachloride(82), "Quantum Biochemistry"(134)
1964 -- Role of metal/melanin/free radical interaction in manganese poisoning and other extrapyramidal disorders(76)
1964 -- Isolation and characterization of purified antiinflammatory protein, "Ontosein"(194).
Mid '60's -- Use of catalase in arthritic diseases in France(195)
Late 60's -- Characterization of granulocyte myeloperoxidase system(l02)
1969 -- Isolation and characterization of superoxide dismutase activity of cupreins(262)
Early '70's -- Role of radical mechanisms in spinal cord injury and stroke (17,18)
1972 -- Characterization of NAD(P)H oxidase system of granulocytes(5)
1972 -- Ontosein (a.k.a.,Orgotein,SOD,Cuprein,Palosein)approved as veterinary antiinflammatory agent in US.
Methods of Detection of Active Oxygen and Free Radical Species.
The high reactivity of free radicals and active oxygen species virtually precludes direct detection in organs and tissue by electron spin resonance techniques. In order for specific species to be detected in aqueous systems, micromolar steady-state concentrations are required.
Such levels are rarely reached in vivo .since the life span of radical species is usually extremely short, I msec. or less. Other authors (27,28) have reviewed the various techniques for the detection of radical and related species in biological systems. Table II outlines some of these methods: Such techniques may take advantage of the relative specificity of such enzymes as SOD or catalase toward active species of oxygen, involve the inhibition of a process by antioxidants or measure production of specific reaction products.
Radical production can also be measured by means of "spin-trapping" techniques, in which a biologically-produced short-lived radical oxidizes another compound to form a long-lived (and thus detectable) radical derivative.(27,28)
Active Oxygen System.
While other radical species may be of importance in such processes as drug metabolismll.(15,20.24-30.34,42,4755.76-89) and in radical chain reactions,(8-19) radical forms of oxygen are the penultimate species in aerobic systems. Figure 3 presents a simplified outline of the major steps in the four-electron reduction of oxygen to water, which occurs through the addition of single electrons to oxygen within the cell by enzymes or by simple chemical reducing agents. (1-42,90-93) Other, less-defined, species (such as hydroperoxides) may also be present.
Mitochondria carry out the entire four electron reduction of molecular oxygen to water at cytochrome oxidase. Likewise, the cytochrome P-450 centered mixed function monooxygenase of liver endoplasmic reticulum - and of other tissues - reduces molecular oxygen to active metabolites, which it uses to oxidize a wide variety of substrates.
Although the chemistry involved is beyond the scope of this review [for further information the reader is referred (034.81-83.92], the reaction occurs by a series of single-electron transfer steps. The products include free radical intermediates, which are subsequently either reduced further or released from the active site of the enzyme and react with nucleophilic groups in the immediate molecular environment - including molecular oxygen.(8,9.11,15.24,27.28.30) Similarly, xanthine oxidase4 oxidizes products of purine catabolism as well as other oxidizable substrates with the generation of reactive oxygen metabolites, while vitamin K-dependent carboxylation of clotting factors in liver also involves generation of active oxygen species(93).

The NAD(P)H oxidase ("superoxide synthetase")/ myeloperoxidase system of inflammatory cells (5,6,3l-33,3S-40,46,94-106) is also of g!eat importance. This membrane-bound system, which is activated by the phagocytosis of foreign materials such as bacteria by phagocytic white blood cells, generates large amounts of superoxide and other microbiocidal and cytocidal oxygen derivatives (Figure 4). Similar systems may also be present in T -lymphocytes, 96 platelets, 10,97 conjunctival mucus (98) and adipocytes.(61). Electrons may also be added directly to molecular oxygen in "autooxidation" reactions such as those involved in the typically metal-catalyzed direct transfer of an electron from a reducing substrate to molecular oxygen or peroxide without an enzyme mediator. (16-21,24,25,52,107-1 17) Reducing agents participating in such reactions include catechols, . .d 24 lfh d I d 24107-109 unc aCl, su y ry compoun s, , quinones, 110 metals/4,3s,1 10-1 16 halides/4 alpha and beta chains of hemoglobin,Il6,117 ascorbate/4,S7,114,IIS paraquat,44,63 and anthracycline drugs such as adriamycin.2o,2s,42,sl If the oxidized derivative is then re-reduced by some other agent such as NADPH, a process known as "redox cycling" occurs. Such a mechanism has been proposed to explain the toxicity and action of quinone derivatives of adriamycin [reviews/O,2S,42,SI] which may shuttle electrons between the hexosemono-phosphate [HMP] shunt and molecular oxy-gen (Figure 5).
Singlet Oxygen and other Excited Molecules. An excited-state species is produced when an orbital electron absorbs energy
FIGURE 3.
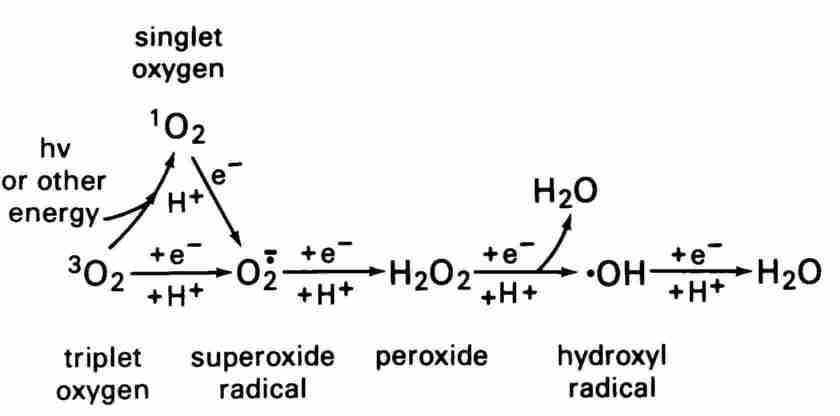
The active oxygen system. In biologic systems molecular oxygen is reduced to water in four oneelectron steps. Reduction of molecular oxygen to superoxide, and of peroxide to hydroxyl radical are "spin forbidden" and thus are slow unless catalyzed by a heavy ion. Alternative spin-permitted pathways for the reduction of 02 include interaction of molecular oxygen with the excited triple state of another molecule to produce singlet oxygen (from light (7,91,118,119) or an excited state molecule (99120121) and jumps to a higher energy orbital on the same atom. Excited-state species produced may be highly reactive and participate in reactions not unlike those of free radicals.(91,99,119-122) Singlet oxygen (Figure 2), itself a strong oxidizing agent, may be responsible for some of the effects assigned to other active oxygen species such as superoxide.(91,122) Roles for singlet oxygen have also been postulated in photosensitized reactions,(90,91,113,119) and in antimicrobial defense. (5,6,33,46.99, 105,120).
In biological systems the sources of the electrons are generally enzymes (e.g., NAD(P)H oxidase) and reducing substances (electron-donors). Simplistically, electron-donors act as antioxidants by (e.g.) reducing more reactive species such as trichloromethyl, superoxide, or hydroxyl radicals to less reactive species such as chloroform, peroxide, or water. Conversely, electron-donors act as pro-oxidants by reducing less reactive species such as molecular oxygen and peroxide to more reactive species via reactions which are typically mediated by the cyclical reduction/oxidation of transition-metal ions. The reduction of peroxide to hydroxyl radical by ferrous iron is known as Fenton's reaction."2 Peroxide and superoxide can also react in the presence of a metal ion to produce hydroxyl radical and molecular oxygen. This latter reaction is called the "Haber Weiss Reaction". (111-115).
Cellular Defense Against Radical Species.
The ubiquity and reactivity of radical-generating systems in cells has resulted in the evolution of defense mechanisms against the damaging effects of such powerful oxidizing agents.(4,12,16-19,21,36,123-128) Figure 6 summarizes the activity of these intracellular enzymes.SOD (436 125 126). Superoxide dismutase and catalase (1-4,126) catalyze the dismutation of superoxide and hydrogen peroxide, respectively, .GSH peroxidase reduces hydrogen and organic peroxides (123,124,126) to water and alcohols, respectively. GSH S-transferases (27) act by transfering glutathione residues to electrophilic reactive metabolites of xenobiotics.
Oxidized glutathione (GSSG) produced is rapidly reduced by reactions utilizing NADPH generated from various intracellular systems, including the hexose-monophosphate shunt.(123,124). Various organelle specific isoenzymes of superoxide dismutase exist. (4,125). The Zn, Cu SOD is cytoplasmic, while the Zn, Mn enzyme is chiefly mitochondrial. Neither isoenzyme is found in high concentrations in extracellular fluids. (4,36,125,129).
Many reducing agents (Table III) also serve as antioxidants, reducing the more active
radical species such as peroxy or hydroxyl radicals (1,3, 14-21,24,58, 130-134) to less reactive forms (e.g., water) (Figure I), as well as de-excite singlet oxygen.(ll9,l35) These compounds also terminate radical chain reactions.(14-21,24).
However, chemical antioxidant defense is a double-edged sword. First, when a reducing agent scavenges a radical, its own radical derivative is formed. Thus, unless the radical is extremely stable, a radical chain reaction may continue. Second, as implied in Figure 2, a reducing agent may itself reduce oxygen to superoxide or peroxide to hydroxyl radical in autooxidation reactions(20,24,107-117. For example, ascorbate (57,114) and uric acid (4) which may functlion as antioxidants, parhcIpate in autooxidations, either directly, by reducing some other oxygen activator such as a transition-series metal or quinone, or by acting as an enzyme cofactor (19,24,57,58,107-117)

FIGURE 6. Antioxidant defense - enzymes. Three important intracellular enzymes constitute antioxidant defense; superoxide dismutase (SOD), catalase, and the GSH peroxidase/GSSG reductase system. SOD catalyzes the dismutation of superoxide, catalase the conversion of hydrogen peroxide to H2O and O2, while GSH peroxidase transfers electrons from GSH to reduce peroxides to water. The oxidized glutathione produced (GSSG) is re-reduced back to GSH by glutathione reductase utilizing NADPH produced by the HMP shunt.acting as an enzyme cofactor(19,24,57,58,I07-117.
Such processes may account for ascorbate's ability to depolymerize DNA,(57) inhibit brain N a + / K+ ATPase, (114) potentiate the toxicity of paraquat,(136) and mediate lipid peroxidation.(1l4) They may also contribute to the pathophysiology of disorders of purine metabolism. (24,26,137,138). The exact mix of pro or antioxidant properties for a particular reducing agent is a complex integration of several factors. In the case of hydroxyl radical scavengers, the product of radical interaction with the antioxidant is generally much less reactive than hydroxyl radical, one of the most powerful oxidizing agents known. Or, the radical forms may be sufficiently stable and present in high enough concentration that they self-annihilate - as is the case with glutathione and superoxide. Similarly, pH greatly influences the direct reduction of oxygen to superoxide by sulfhydryl compounds, (107) while other local factors such as the molar concentration of oxygen molecules are also important.
Excited-state derivatives such as singlet oxygen and the excited triplet (diradical) states of other molecules may be quenched by interactions with conjugated diene systems such as those found in carotenes,(1,19), tocopherols,(135), or the melanins. (24,26,45, 73-79). As is the case with reducing antioxidants, such compounds may also produce active electronic species (45,73-79,118) and perhaps disease.
TABLE III. Antioxidant defense - chemicals (modified after refs. 17-19)
Ascorbate
Tocopherols
Purines (esp. uric acid)
Sulfhydryl compounds (e.g., cysteine, homocysteine)
Catechols Monophenols (e.g., tyrosine)
Reduced gluthatione (GSH)
Selenium Carotenes
folates Corticosteroids
other sterol derivatives
Superoxide (?)
Mannitol (*OH Quencher)
Caeruloplasmin
The Role of Active Species in Pathological Processes.
In addition to environmental causes such as oxygen(4,13,29,139,147), light (90,91), or ionizing radiation (53,148-149) three physiological circumstances result in extraordinarily high local fluxes of radical species: (1) activation of the P-450-centered mixed function oxidase systems of endoplasmic reticulum, (2) activation of NADPH oxidase in phagocytes in response to antimicrobial defense and inflammation (Figure 4) and (3) the presence of extraordinarily high levels of compounds which can reduce oxygen directly in autooxidation reactions.
Under such circumstances, the rate of active species generation may exceed the local capacity of the antioxidant defense and may contribute to injury.
However, before we proceed we must again insert a note of caution. With the exception of melanin, the radical species present in biological systems are very short-lived and are present at such low concentrations that they are usually not detectable in vivo by electron spin resonance spectrometry. Indeed, they may be detected in vitro only under very special conditions (27,28). Thus, evidence for the involvement of any radical or excited-state species in a particular biological process (much less in a human disease) is necessarily indirect, often circumstantial and must be taken cum grano salis (with a grain of salt). Nonetheless, multiple bits of evidence (which taken separately are inconclusive) collectively strongly support a role for electronically-activated species in at least some processes such as inflammation and in the toxicity of some drugs, although the exact role and identity of such species is a subject of much controversy (e.g. 150,151).
Table IV outlines some criteria for acceptance of a role for free radicals in specific human diseases. These criteria include such things as inhibition by a presumably specific enzyme such as SOD or the detection of specific reaction products. For example, Wilson's Disease (with high local tissue copper contents, and lower levels of ceruloplasmin, a copper containing protein which may function as an extracellular antioxidant/I,128) qualifies as a putative radical-associated disease under criteria I and IV. Yet, to the best of our knowledge, no one has yet demonstrated an accelerated flux of superoxide, increased products of lipid peroxidation or other free radical metabolites in this disease. Further application of these criteria to other disease processes is an exercise for the critical evaluator to ponder.
TABLE IV. Criteria for involvement of radical processes.
I. The condition is known to be associated with abnormal production of free radicals and other electronically activated species (e.g., granulocyte activation in inflammation, or the presence of a putative charge-transfer agent in abnormal amounts or forms - such as copper in Wilson's disease).
II. Demonstration of specific radical species or their unique reaction products at the site of a lesion.
III. In vitro demonstration that radical species are involved in important mechanisms relevant to the disease in question.
IV. Production of similar symptoms and lesions by otherwise dissimilar chemicals which produce free radical species in common, or which inhibit or deplete components of the natural antioxidant defense system (the "common symptom" test).
V. A corollary to IV: the ability to modulate the pathophysiologic progression of a disease pharmacologically through the intervention of ectopically administered SOD, catalase, antioxidants, or free radical quenchers.
Inflammation.
The enzymatic production of active oxygen species by inflammatory cells (Figure 4) may contribute to the pathophysiology of leucocyte dependent inflammatory processes [reviews l,7,16,31-33]. As outlined in Figure 4, in addition to their direct action on cellular constituents (1,7,16,31-33,38-41,96,100,152) oxygen metabolites may also act as specific modulators of the inflammatory process. For example, in vitro active oxygen species can affect the activity of inflammatory immunomodulators such as interferon (153) leucocyte-dependent inflammatory processes (Reviews, 1,7,16,31-32) (154-156), leucocyte clastogenic factors (154-156) , lymphocyte clastogenic factors(157-159), soluble immune response suppressor (160), serum protease inhibitors (161-164), and vascular permeabi1ity-regulating factors.(165,166). Similarly, extracellular active oxygen species may also directly influence platelet (l67-I71) and fibrocyte (63,172) function. They may also be involved in the metabolism and action of such important modulator substances as the prostaglandins (41,68) leucotrienes,(17I), unsaturated fatty acids (41,68-71) and cyclic nucleotides (61-67). They may also influence interactions between lymphokines and macrophages (l56,173) and lymphocyte function (l74) as well as induce histamine release from mast cells (175). The specificity of such effects suggests that active metabolites of oxygen may be acting more as messenger substances than as non-specific chemical reagents. (emphasis-added).
Such circumstantial in vitro evidence hould be assessed with the special caution reserved for this difficult field. Nonetheless, taken globally, it is not surprising that antioxidents (l76-183), SOD (1,138,154,165,177,184-194) and catalase (32,177,195) should ameliorate inflammatory symptoms in human and animal systems.
This is of some clinical importance, since an antiinflammatory pharmaceutical preparation rich in SOD ("orgotein") is used in veterinary medicine and recently has been shown to be both effective and apparently safe in the treatment of various inflammatory lesions in man. (1,138,154,165,177,184-194). Catalase has also been used in the treatment of arthritic disease in man with reported success. (195). Likewise, the action of such established antiflammatory drugs as the corticosteroids, (196) penicillamine,(197) and many non-steroidal antiinflammatory agents (1,76-183,198-200) may be at least partially dependent upon interference with active oxygen metabolism in phagocytes.
Further, peroxidase release from eosinophils may playa similar role in inhibition of the inflammatory response (201) while the antioxidant properties of ceruloplasmin may also give this compound antiinflammatory properties.(21,128).
An illustrative (albeit circumstantial) model for the role of superoxide in rheumatoid arthritis can be postulated as follows: Granulocytes tend to be concentrated at sites of active rheumatic disease, presumably in response to the presence of immune complexes and other immunomodulator substances. Superoxide, peroxide, and other active oxygen species produced further kindle the inflammatory process by specific mechanisms such as those outlined in Figure 4. Catalysis of radical oxidations by transition-sernies metals may also play a role. Agents such as ectopic SOD may interfere with this process by destroying active oxygen species or increasing peroxide fluxes, thus interfering with one or more of the mechanisms detailed above and in Figure 4.
Since SOD is one of the most substrate-specific enzymes known, at first sight its efficacy in rheumatoid arthritis and other lesions strongly implies a role for active oxygen species in such diseases and a role for superoxide-destroying agents in their treatment. However, there are serious problems with such an assumption. For example, the course of action of SOD as an inflammatory agent often bears no apparent relationship to its serum levels (1,185) and the "denatured" enzyme still possesses significant antiinflammatory properties. 185 Feel (51) even ably questions the specific role of the protein in destroying superoxide.
With this caution, active oxygen species may play a significant role in the etiology of other inflammatory lesions in man, For example, SOD is reported to be effective in the treatment of lupus erythematosis,(43,158) and unique light-activated, superoxide-dependent lymphocyte clastogenic factors present in the serum of patients with lupus and other collagen diseases may account for some of the photosensitivity of this group of disorders.(157-159). Both direct and indirect production of active oxygen species may also have a role in the pathophysiology of gout and other hyperuricemic syndromes.(24,26,173,138,203,204). For example, urate, a reducing agent, is present in the extracellular environment at concentrations approximating 0.3 mMolar (130). Like many reducing agents, it apparently has both antioxidative (3O-134) and autooxidative (24) properties. In fact, it may have taken over some of the functions of ascorbate in primates.(133). Likewise, xanthine oxidase is an effective producer of oxygen radical species, while urate itself stimulates the production of active species of oxygen by phagocytes (203,204) and may protect cycloepoxigenase from autooxidation.(131) The effectiveness of SOD in the treatment of urate-induced inflammatory disease in Dalmations suggests a role for superoxide in this lesion.(138).
Production of active oxygen species by activated phagocytes may also have a role in vascular (and other) damage following endotoxin shock,(144,145,205), burn-induced plasma volume loss (71) and even in atherosclerosis.(205). Similar mechanisms may account for the possible role of radical species in the progression of damage following neuronal injury.(14,17,56,171,206). Antioxidants such as the methoxyphenols are apparently effective in the amelioration of both experimental cerebral edema and spinal cord injury.(17,56). However, once again we must emphasize that, like most else in this field, the evidence for free radical involvement in inflammation and neuronal injury is circumstantial and has not been proven conclusively.
Radical Mechanisms in Drug Toxicity.
Oxidation of chemicals by the p450 system generates free radical and other metabolites such as epoxides and aldehydes which may interact with cellular constituents. (8,9,11,15,1721,27,28,30,34,73,80-86,110,207,208) F I t. Forr example, partly as a result of recent spin-trapping studies,(8,9,73) perhaps the best established role of free radicals per se is in the toxicity of halogenated hydrocarbons such as carbon tetrachloride and halothane. (8,9, 11,28,30).. Nonetheless, (typical for this field) it remains unclear whether it is (e.g.) the trichloromethyl radical, peroxide, or the peroxy radical or its reaction products with oxygen which cause injury in carbon tetrachloride poisoning.
The P-450 centered mixed-function oxidase can also synthesize simple and complex quinones, imine quinones, and nitro aromatics from a variety of aromatic xenobiotics.(27,28,34,73,80-86,110,207,208). In turn, these metabolic products can undergo cyclic one-electron oxidationj reduction reactions2° which ultimately reduce molecular oxygen to superoxide (Figure 5).
Thus, the toxicity of acetaminophen may be due to its oxidation to an iminequinone, which in a reduced state may either interact with vital nucleophilic targets within the cell, or generate superoxide anion, or both.(81,85), Similarly, the antibacterial and toxic potential of 5-nitrofurantoin appears to be related to its reduction to an anion radical, which among other alternatives can generate superoxide anion.(83). Similar mechanisms have been proposed to account for toxicity of chlorpromazine.(27,28,77,80). Also, the phototoxicity of many agents appears to depend upon the production of singlet oxygen in the presence of an excited triplet state produced by the action of light on the photosensitizer.(37,90,91).
Such reactions follow similar pathways to ultimately result in the reduction of oxygen to water. Light-induced superoxide production may be responsible for the photosensitivity found in lupus and other collagen diseases. (157-159), Direct, nonenzymatic reduction of oxygen to active species may have a role in both the action and toxicity of the antitumor agents adriamycin (I,20,22,25,42,51,52,67), bleomycin (22,49,51, 53b-55) and cis-platinum.(1,49.51). In vitro, both . .adriamycin and bleomycin' (otherwise totally-dissimilar drugs) bind to DN A and damage it by active-oxygen dependent mechanisms and may participate in redox cycling.(20,42)
Likewise, their toxicity in .many systems, and that of cis-platinum, (1,49,51,209) can be ameliorated by the administration of ectopic SOD and/or catalase. Vitamin E ameliorates the toxicity of adriamycin (5) but only if given before administration of the antitumor agent.(210), Significantly (although it may not be generally true in man (211), many animal tumors appear to be deficient in defense mechanisms against radical species relative to normal cells (review, 22). This may increase the relative antitumor specificity of radical-generating antitumor agents(22).
Since SOD and catalase (which probably do not penetrate into cells) ameliorate the toxicity of such agents in vivo, a significant fraction of their toxicity may be due to extracellular production of oxygen metabolites, in line with the relative paucity of defense mechanisms in the extracellular space. In support, adriamycin is cytotoxic in vitro when bound to an insoluble extracellular substrate.(212,213). In turn, this holds out the possibility that the (putative) intracellular antitumor activity of such drugs (say, at the DNA level) may be separable from that fraction of their toxicity which is extracellular. SOD apparently does not diminish the antitumor effect of such drugs.(1,51). However, the situation is confused by the apparent partial antitumor activity of SOD itself, (43,51,215) which may happen with the "denatured" enzyme,216 and which is roughly equivalent to cis-platinum in at least one experimental animal tumor system. (57).
Active Oxygen and Fibrosis.
Oxygen in high concentration produces diffuse alveolar damage (DAD), often progressing to interstitial pulmonary fibrosis, which is essentially identical to that caused by a myriad of other causes, including drugs, endotoxins, thermal . .injury, and infammatory processes, , , ,(146,217,218), many of which may involve production of active oxygen metabolites. The toxicity of oxygen itself is likely due to the productlon of active oxygen species, while the radical-producing antitumor agent .bleomycin produces pulmonary injury and subsequent interstitial pulmonary fibrosis.(146). Further, a variety of other radical generating agents are associated (perhaps fortuitously) with similar alveolar damage and/ or fibrotic symptoms. Examples include the pulmonary lesions induced by such agents (44, 219) as paraquat,' mtro furantoins, or bleomucn and the vitreous fibrosis induced by interocular hemoglobin or elemental copper. (220).
Significantly, ectopic SOD (221) and catalase (222) apparently give some protection in experimental paraquat poisoning, although this has not as yet been borne out in human trials.(44). Likewise, SOD has been reported to be effective in such fibrotic lesions as Peyronies disease (l89) and radiation-induced cystitis.(148,188). Superoxide is reported to activate fibrocyte function in vitro, stimulating such processes as collagen production (l72) and guanylate cyclase.(63).
We speculate that the stimulation of (e.g.) the fibrotic response by these radical-generating agents may be a consequence of their common interaction with existing modulator mechanisms coupling production of active oxygen species by inflammatory cells with fibrocyte activation, collagen production, and wound healing as part of the inflammatory response.
Other Charge- Transfer Agent Associated Diseases.
The chronic presence of elevated levels of other autooxidation-catalyzing charge-transfer agents such as copper or manganese is associated with human disease states, although it is again an open question whether radical mechanisms contribute to the pathophysiology of such diseases. Nonetheless, some clinically useful (if speculative and unproven) associations have been made.
For example, the conjectures of Cotzias et al.(76.223) concerning the possible role of a metal/ neuromelanin/ free radical interaction in the pathophysiology of manganese poisoning, Wilson's Disease, and phenothiazine toxicity were one of the major factors leading to the trial of levo-dopa in manganese poisoning and, ultimately, Parkinson's disease.(223) Similar considerations concerning inner ear melanin, drug-induced deafness, and nephrotoxicity prompted the use of SOD and catalase in the amelioration of cis-platinum toxicity.(49.5l). Too broad an area to consider here in detail, various aspects of this field have been the subject of work by other authors e.g,, (24.26.45,75-80,89,223-250).
Briefly, ionic forms of copper, iron, and manganese (as well as other "charge transfer" agents) can all catalyze radical oxidations in vitro. The respective diseases associated with chronic elevated systemic levels of such compounds (e.g., Wilson's disease, hemochromatosis and chronic manganism) are variably associated with one or more of a set of six specific symptoms. These symptoms may include movement disorders, "psychosis", pigmentary abnormalities, deafness, fibrotic lesions such as cirrhosis, and arthritic symptoms. For example, hemochromatosis, an iron storage disease, may present with most or all of these symptoms (vis, the role of iron in catalysis of radical oxidations (3,53-55,92,lll-115).
As we have seen, the last two symptoms (fibrosis and arthritis) may be referable to radical mechanisms under other conditions (Figure 4), while several putative neurotransmitter-dependent processes (such as burn-induced plasma volume loss,(71) oxygen, (139,143) or adriamycin toxicity (51) serotonin release from platelets (168), histamine release from mast cells,(175) and autocoid-induced edema in mice (l65)) may be modulated by active species of oxygen. Direct oxidative pathways for neurotransmitter metabolism are also possible (4,84,88,89,114,244,246-250) as are other effects on neurotransmitter function. (17,24,26,45,49,89,1 14,143, 223,249).
The same associations between pigmentary abnormalities and other specific symptomatology appear to hold for other "chargetransfer" agent associated diseases such as alcaptonuria, hyperruricemic syndromes, homocystinuria, and chronic iodism.(24,26,138,235). Further, both drug-induced cutaneous photosensitivity (a process which likely involves generation of active oxygen species (90,91)), and the clinical use of such oxygen-radical-generating antitumor agents as bleomycin and adriamycin (253) are commonly associated with hyper-pigmentation. While it is not within the bounds of this review to consider such associations in depth, many charge-transfer agents bind readily to melanin (229,230) for much the same reason that they bind to molecular oxygen to catalyze autooxidations (24,26). Further, since melanin may be a protective agent against active species, the common presence of pigmentary abnormalities in such diseases and in inflammatory and photosensitivity diseases (e.g., Lupus erythematosus) may be an outward reflection of some ongoing electronically-activated process. Once again, such circumstantial evidence must be approached with caution.
Interestingly, pigmentary abnormalities alone may be associated with specific neurological symptoms such as deafness and movement disorder.(26). For example, the association between pigmentary abnormalities and deafness in such diseases as Waardenburg's and Usher's syndromes is a commonplace in audiology (228-232) and may be present in over 10% of cases of severe congenital deafness.(231). Likewise, the toxicity of such oxotoxic/ nephrotoxic drugs as kanamycin (26,45,47,49) and cis-platinum (49,51,149) may be due to free radical mechanisms and / or to binding to inner ear melanin.(229,230) Similar mechanisms may hold for the nephrotoxicity of agaricus bisporus toxin (254) and para-aminophenol,(110) as well as the renal toxicity of radical-producing agents such as adriamycin or paraquat.
Enzyme Deficiencies.
The various protective mechanisms are usually adequate to protect biological systems against normally present active species except under conditions of high radical load. However, in glucose-6phosphate dehydrogenase (G6PD) deficiency the activity of the GSH reductase/ peroxidase system of the red blood cell is greatly curtailed secondary to nonregeneration of NADPH (Figure 6). The red cell is particularly subject to radical load because the hemoglobin and its side-chains may be generators of active oxygen species (117).
Normally, even in the absence of G6PD, the remaining activity of the GSH reductase/ peroxidase system in concert with the other protective mechanisms is adequate to protect the red cell. However, under conditions of oxidative load as may occur in antimalarial treatment, they are inadequate, resulting in hemolysis. (101,124,255). Acatalasemia in humans has minor symptomology (sterile mouth ulcers (256), perhaps due to the intactness of other defense mechanisms. Similarly, a relative deficiency of granulocyte superoxide dismutase has been reported in juvenile rheumatoid arthritis.(257) Deficiency of the active-oxygen generating NADPH oxidase system in granulocytes is present in chronic granulomatous disease (7,8,13) and may be associated with sepsis in burn injury.(258).
Summary and Clinical Implications.
In summary, we must reiterate a few key points. First, most evidence for active radical mechanisms in biological processes and in human disease states is circumstantial. At present we are unable to measure such species directly in man. Clearly, as our understanding of the problems increases, better techniques for the study, diagnosis, and moderation of such processes should develop. Second, protective mechanisms suffice to protect organisms against such species under normal conditions and are adequate enough that endogenously generated active species may be utilized as substrates in (e.g.) antimicrobial defense and xenobiotic metabolism.
TABLE V. Clinical trials with SOD.
SOD in its human pharmaceutical preparation "Ontosein" has been reported to be effective in both double blind placebocontrolled and open studies covering a variety of human diseases. While these findings are supportive of a role for superoxide radical in the etiology of such diseases, they are certainly not conclusive evidence. SOD may be acting completely independently of its ability to destroy superoxide radical, for example, by increasing peroxide fluxes"'" 49,51,139 or even by acting directly as an immunomodulator (153).
Double- Blind. Placebo- Controlled Studies:
Rheumatoid Arthritisl Duchenne Muscular Dystrophy (65). Radiation-Induced Cystitis (l48,188)
"Open" Studies
Osteoarthritis (1, 185, 190,193)
Interstitial Cystitis (191)
peyronies Disease (189)
Myelosuppression from cancer chemotherapy (187) Others (41,185,190,191)
Thus, acute free radical pathology should occur under conditions of extraordinary radical flux. These include inflammation, radiation or oxygen therapy, metabolism of specific xenobiotics, or overload of autooxidationcatalyzing agents such as manganese, copper, or iron. Further, as implied in Figure 4, oxygen radical production may be a fundamental mediating component of the secondary systemic response to injury ("inflammation") from whatever source, be it (e.g.), thermal injury, drugs, or infection.
This apparent role of oxygen metabolites in the final common pathway of the secondary response to tissue injury even further confounds the exact role of such species in pathogenesis. For example, are the ameliorative effects of SOD or catalase on the toxicity of radical-producing antitumor agents such as adriamycin or radiation due to the inhibition of the primary toxic action of such agents or to inhibition of the secondary systemic response to such injury? We note that SOD may be protective even in the case of antitumor agents for which there is no evidence of radical production. (187)
Of great importance is providing a basis for understanding present and potential therapy. Orgotein ("Ontoseinr'), the pharmaceutical preparation of SOD, has long been used in veterinary medicine as an antiinflammatory agent under the trade name "Palosein'(168-169). Indeed, orgotein was used as an antiinflammatory agent well before it was realized it was SOD! (Table I). This agent has recently completed clinical trials for a variety of diseases (Table V) and currently awaits approval for human use in the U.S. Presently its clinical use is permitted in Europe and Japan. Perhaps the most noteworthy aspect of the pharmaceutical SOD preparation is its low order oftoxicity. If ectopic SOD lives up to its expectations, it will be a welcome addition to the therapeutic armamentarium, perhaps partially replacing steroids in long-term therapy of immunologically-based diseases such as lupus or rheumatoid arthritis.
Potential applications of free radical research include the use of catalase or catalase-like agents in (e.g.) inflammatory lesions,(195), oxygen toxicity,(139), burn-induced plasma volume loss, paraquat poisomng and immune injury to the lung.(217). Similarly, SOD, catalase and other antioxidants apparently ameliorate the toxicity of antitumor chemotherapeutic agents and could be used as adjuvants to radiation therapy; while increased understanding of the molecular basis of the toxic potentials of other agents such as halothane and amino glycoside antibiotics should result in ways to abrogate their toxicity. Finally, the emerging picture of radical mechanisms as fundamental common components of many basic pathophysiologic processes (such as in cerebral edema, parkinsonism, and pulmonary disease) clearly has clinical implications far beyond inflammation and drug toxicity.
Glossary
Auto-oxidation. A non-enzymatic oxidation-reduction reaction involving two or more chemical compounds one of which is usually oxygen or one of its active species.
Charge-transfer agent. Any compound which can easily donate or accept electrons.
Diamagnetic. Compounds with pairs of orbital electron(s) with paired spins, such that the molecule has no net magnetic moment.
Dismutation. An oxidation-reduction reaction involv-
ing a single chemical compound.
Free Radical. An atom or molecule which has one or more electron(s) with unpaired spin(s).
Paramagnetic. Compounds with an orbital electron of unpaired spin having a net magnetic moment.
Reduction/ Oxidation - (Redox). Respectively, gain or loss of electrons by an ion or molecule.
Singlet state. State in which all spin-derived magnetic fields are paired. Ground state of most molecules, but excited state of oxygen. The form of 102 responsible for most of its biological activity is the "Delta" form, a potent electrophile.
Spin - Classically, the magnetic property of the electron derived from its nature as a "spinning" negative charge analogous to a current flowing in a "ring". Thus, electron may be viewed as a bar magnet with its north pole oriented either "up" or "down".
Triplet (diradical) state - Species with even number of electrons,. but with two unpaired electrons of parallel spin in separate orbitals. Usually excited state, but is the ground state of oxygen.
Keywords: nitrone nitroxide cerovive (tm) nxy-059 centaur astrazenica renovis stroke ischaemic injury pbn
REFERENCES
I. Huber, W., In: Inflammation-Mechanisms and Treatment ed: P. A. Willoughby and J. P. Giroud.
University Park Press, Baltimore, p. 27 (1980).
2. Bors, W., Saran, M., Lengfelder, E., Spotte, R. and Michel, C. Curro Top. Radia. Res. Q. 9:247 ( 1974).
3. Willson, R. L., In: Oxygen Free Radicals and Tissue Damage. Ciba. Symp. 65 (New Series), Amsterdam, Exerpta Medica p. 19 (1979).
4. Fridovich, I., Bioscience 27: 462 (1977).
5. Babior, B. M. N. Engl. J. Med. 298:659 and 721 (1978).
6. Klebanoff, S. J. Semin. Hematol. 12: 117 (1975).
7. Kuel, F. A., Humes, J. L., Torchiana, M. L., Ham, E. A., Eagan, R. W. In: G. Weissman, B. Samuelson and 8. Paoletti, eds. Adv. in Inflammation Res.. New York, Raven Press, p. 419 (1979).
8. Reynolds, E. S. and Moslen, M. T., In: Free Radicals in Biology Vol. 4. ed. D. A. Pryor, Academic Press, New York, p. 49 (1980).
9. Reynolds, E. S., Moslen, M. T. and Treinen, R. J. In: Oxygen and Oxy- Radicals in Chemistry and Biology. New York Academic Press, p. 169 (1981).
10. Marcus, A. J., Semin. Hematol. 16. 188 (1979).
11. Mason, R. F. and Ching nell, C. F. Pharmacological Reviews 33:189 (1981).
12. N. M. Emanuel. Quarterly Reviews of Biophysics 9:283 (1976).
13. Deneke, S. M., Fanburg, B. L., N. Engl. J. Med.303:76 (1980).
14. Demopoulos, H., Flamm, E., Scligman, M. L., Power, R., Pietronegro, D. and Ransohoff, J. In:
Oxygen and Physiological Function. ed.: F. F.Volsis, Professional Information Library, Dallas, p. 491 (1977).
Pso, P.O., W. J. and Lorentzen, In: Free Radicals in Biology. Vol. 3. ed. W. A. Pryor, Academic Press, New York, p. 251 (1977).
Delmaestro, Rolando F. Acta. Physiol. Scand.Suppl. 492:153 (1980).
17 Demopolous, H. B., Flamm, E.S., Pietronegro, D.D., and Seligman, M. L., The free radical pathology and the microcirculation in the major central nervous system disorders. Acta. Physiol. Scand.Suppl. 492:91 (1980). Also see***
18. Demopolous, H. B., Fedn. Proc. 32:1859 (1973).
19. Pryor, W. A., Photochem. Photobiol. 28:787 (1978).
20. Kappus, H. and Sies, H. Experientia 37:1233 (1981).
21. Dormandy, T. L. Lancet. ii:647 (1978).
22. Oberley, L. W., and Beuttner, G. R. Cancer Research 39, 1141 (1979).
23. Borg, D: C. In: Free Radicals in Biology., ed. W. A. Pryor, Academic Press, New York, p. 69 (1978).
24. Proctor, P. H. Physiol. Chem. Phys. 4:349 (1972).
25. Olson, R. D., Boerth, R. c., Gerver, J. G., and Nies, A. S. Life Sciences 29:1393 (1981).
26. Proctor, P. Pigment Cell, 3:378 (1976).
27. Kalyanaraman, B. Rev. Biochem. Tox. 4:73 (1982).
28. Pryor, W. A. In: (ed.) Bhatnager, Molecular Basis of Environmental Toxicity, Ann Arbor Science publishers, Ann Arbor, p. 3 (1980).
29. Rinaldo, J. E., and Rogers, R. M. N. Engl. J. Med.306:900 (1982).
30. Farber, J. L. Lab. Invest. 47:114 (1982).
31. Weiss, S. J., and LoBuglio, A. F. Lab. Invest. 47:5 (1982).
32. Fantone, J., and Ward, P. A. Am. Journal of Pathology. 107:396 (1982).
33. Klebannof, S. J. Ann. Int. Med. 93:480 (1980).
34. Sirica, A. E. and Pitot, H. C. Pharmacological Reviews, 39:205 (1979).
35. Hochstein, P., Kumar, K. S., Forman, S. J. In: The Red Cell, A. R. Liss, New York, 669 (1978).
36. Fridovich, I. In: Pathology of Oxygen, Autor, A.P., ed., Academic Press, New York, p. 1(1982).
37. Foote, C. S., In: Pathology of Oxygen. Autor, A.P., ed., Academic Press, New York, p. 21 (1982).
38. Babior, B. M., In: Pathology of Oxygen. Autor, A.P., ed., Academic Press, New York, p. 45 (1982).
39. Hammerschmidt, D. E., and Jacob, H. S. In:Pathology of Oxygen. ed., A. P., Autor, Academic Press, New York, p. 59 (1982).
40. Oyanagui, Y. In: Pathology of Oxygen, A. P.Autor, ed., New York, Academic Press, p. 99 (1982).
41. Kuehl, F. A., Ham, E. A., Egan, R. W., Dougherty, H. W., Bonney, R. J., Humes, J. L. In: Pathology of Oxygen. Autor, A. P., ed., Academic Press, New York, p. 175 (1982).
42. Doroshow, J., and Hochstein, P. In: Pathology of Oxygen. Autor, A. P., ed., Academic Press, New York, p. 245 (1982).
43. Michelson, A. M. In: Pathology of Oxygen. Autor, A. P., ed., Academic Press, New York, p. 277 ( 1982).
44. Fairshter, R. D., Arch. Intern. Med. 145:1121 (1981).
45. Proctor, P., McGinness, J. and Corry, P., J. Theor.Biol. 48:19 (1974).
46. Allen, R. C., Front. Bioi. 48:179 (1979).
47. Pierson, M., and Moller, A. R., Hearing Research.4:79 (1981).
48. Trager, W. F., Drug Metabolism Reviews. 13:51 (1982).
49. McGinness, J. E., Proctor, P. H., Demopolous, H.B., Hokanson, J. A. and Kirkpatrick, D. S., Physiol. Chem. and Physics. 1:267 (1978).
50. Kahl, R., Trends in Pharmacological Sciences. 3:72 (1982).
51. McGinness, J. E., Proctor, P. H., Van, N. T., Hokanson, J. E., and Kirkpatrick, D. L. In:Pathology of Oxygen, ed., A. P. Autor, Academic Press, New York, p. 191 (1982).
52. Goodman, J. and Hochstein, P., Biochem. Biophys. Res. Commun. 77:797 (1977).
53a. Myers, S. In: Free Radicals in Biology, VoL IV, Academic Press, New York, p. 95 (1980).
53b. Loun, J. W., and Sim, S., Biochem. Biophys. Res.Commun. 77: 1150 (1977).
54. Ishida, R., and Takahashi, T., Biochem. Biophys.Res. Commun. 66:1432 (1975).
55. Yamanaka, N., Masunori, F., Koizumi, K., Nishida, K., Kato, T., Ota, K. In: Tocopherol, Oxygen, and Biomembranes, eds. C. de Dura and O. Haydishi, Elsevier, North Holland Biomedical Press, p. 59 (1978).
56. Demopoulos, H. B., Flamm, E., Seligman, M., and Pietronigro, D. D., In: Pathology of Oxygen, ed., Autor, A. P., Academic Press, New York, p. 127 ( 1982).
57. Morgan, A. R., Cone, R. L., Elgert, T. M., Nucleic Acids Res. 3:1139 (1976).
58. Ascider, K., and Kanamatsu, S., Agri. Bioi. Chem.40: 1891 (1976).
59. Hensiksen, T., Melo, T., Saxebol. G. In: Free Radicals in Biology, ed., W. A. Pryor, Academic Press, New York, p. 35 (1976).
60. McCord, J. M., Science, 185: 529 (1974).
61. Mukherjee, S. P. and Lynn, W. S., Arch. Biochem.Biophys. 184:69 (1977).
62. Mittal, C. K. and Murad, F., J. Cyclic. Nucleotide Res., 3:381 (1977).
63. Vesely, D. L., Watson, B., and Levely, G. S., J.Pharm. Exp. Ther.. 209:162 (1979).
64. Goldenberg, N. D., Graff, G., Haddox, M. K., Stephenson, J. H., Glass, D. 8., and Moser, M.E., Adv. Cyclic. Nucleotide Res., 9:101 (1978).
65. Murad, F., Mittal, C. K., Arnold, W. P., Katsuki, S., Kimura, H., Adv. Cyclic. Nucleotide Res..9:145 (1978).
66. Yoshikawa, K., and Ku{iyama, K., Biochem. Biophys. Res. Comm., 101:927 (1981).
67. Unserferth, D. V., Fertel, R. H., Talley, R. L., Magorein, R. D., and Balcerzak, S. P., Tox. And Appl. Pharm., 60:151 (1981).
68. Kuehl, F. A., Humes, J. L., Torchiana, M. L., Ham. E. A. and Egan, R. W. In: Advances in Inflammation Research, eds., G. Weissman, B.
Samuelson and R. Paoletti, Vol. I. Raven Press, New York, p. 419 (1979).
69. Kuehl, F. A., Egan, R. W., Doughey, H. W., Bonney, J., and Humes, J. L., In: Active Oxygen and Medicine, ed., A. P. Autor, Raven Press.New York, (1982) in press.
70. Kuehl, F. A., and Egan, R. W., Science, 2/0:978 (1980).
71. Hilton, J. E., J. Trauma 20:663 (1980).
72. Sealy, R. c., Felix, C. c., Hyde, J. S. and Schwartz, H. M., Free Radicals in Biology, Vol.
IV, ed., W. A. Pryor, Academic Press, p. 210 (1980).
73. Cope, F. W., Sever, R. J., Polis, 8. D., Arch.Biochem. Biophys. 100. 171 (1983).
74. Mason, H. S., Ingram, D. V. E., and Allen, B., Arch. Biochem. Biophys. 86, 225 (1960).
75. Blois, M. S., Zahlan, A. B. and Maling, J. E., Biophys. J. 4,471 (1964).
76. Cotzias, G. c., Papavasio, Liow, P. S., Van Woert, M. H., and Sakamoto, A., Fedn. Proc. 23: 713 (1964).
77. Forrest, F. M., In: Forrest, I. S., Carr, C. J., and Usdin, E., The Phenothiazines and Structural/yRelated Drugs, Raven Press, New York, p. 255 (1974).
78. McGinness, J. and Proctor, P., J. Theor. Bioi.39:677 (1973).
79. Menon, I. A., and Hakerman, H. F., Brit. J.Dermatol., 97:109 (1977).
80. Forrest, I. S., Forrest, F. M., and Berger, M., Biochem. Biophys. Acta., 29:441 (1958).
81. Thurman, R. G. ad Kauffman, F. c., Pharmacological Reviews 31:229 (1979).
82. Butler, T. c., J. Pharm. Exp. Therap. 134:311 (1961).
83. Thrush, M. A., Mimnaugh, E. G., Ginsburg, E., and Gram, T. E., Biochem. Pharm. 31:805 (1982).
84. Sasame, H. A., Ames, M. M., and Nelson, S. D., Biochem. Biophys. Res. Commun. 78:919 (1977).
85. Mitchell, J. R., Hughes, H., Lauterburg, B. H., and Smith, C. V., Drug Metabolism Reviews 13:539 ( 1982).
86. Naguchi, T., Fong, Kuo-Lan, Lai, E. K., Alexander, S., King, M. M., Olson, L., Poyer, J. L. and McCay, P. B., Biochem. Pharm. 31:615 (1982).
87. Roman-Franco, A. A., J. Theor. Bioi. 97:543 (1982).
88. Borg, D. C., Proc. Natl. Acad. Sci. (USA) 53:829 (1965).
89. Graham, D. G., Tiffany, S. M., Bell, W. R., and Gutkencht, W. F., Mol. Pharm. 14:644 (1978).
90. Foote, C. S. In: Free Radicals in Biology, ed., W.
A. Pryor, Academic Press, New York, p. 213 (1976).
91. Khan, A., J. Phys. Chem., 80:2219 (1976).
92. Gold, A., Toney, G. E., Wisnieff, T. J., and Sangaiah, R., Rev. Biochem. Tox. 4:31 (1982).
93. Sutties, J. W., Trends in Biochemical Sciences, 5:302 (1980).
94. Henderson, W. R., and Kaliner, M., J. Clin.Invest., 61:187 (1978).
95. Walker, L., and Lowrie, D. B., Nature, 293:69 (1981).
96. Roder, J. C., Helfand, S. L., Werkmeister, J., McGarry, R., Beaumont, T. J., and Dueve, A., Nature, 298:569 (1982).
97. Marcus, A. J., Silk, S. T., Safier, L. B., Ullman, H.L., J. Clin. Invest. 59:149 (1977).
98. Proctor, P., Kirkpatrick, D., and McGinness, J., Invest. Ophthal. 16:762 (1977).
99. Rosen, H. and Klebanoff, S. J., J. Bioi. Chem.252:4803 (1977).
100. Slivka, A., LoBuglio, A. R., and Weiss, S. J., Blood, 55:347 (1980).
101. Cox, F. E. G., Nature, 302:19 (1983).
102. Klebanoff, S. J., J. Bacteriol. 95:2131 (1968).
103. Baldridge, C. W., and Gerard, R. W., Am. J.Physiol. /03:235 (1933).
104. Wenning, R. S., Weser, R., and Roos, D., J. Lab.Clin. Med. 85:245 (1975).
105. Beall, G. D., Repine, J. E., Hoidal, J. R., Rasp., R. L., Infection and Immunity, 17:117 (1977).
106. Badway, J. A., and Karnovsky, M., J. Bioi.Cherm., 264:11530 (1979).
107. Misra, H. P., J. Bioi. Chem. 249:2151 (1974).
108. Rowley, D. A., and Halliwell, 8., FEBS Letters 138:33 (1982).
109. Tien, M., Bucher, J. R. and Aust, S. 0., Biochem.Biophys. Res. Cornmun. 107:173 (1982).
110. Calder, I. C., Young, A. C. Woods, R. A., Crowe, C. A., Ham, K. N., and Tange, J. D., Chern. Bioi.Interact. 27:245 (1979).
111. Haber, F. and Weiss, J., Prac. R. Soc. Ser. A 147:332 (1934).
112. Crichton, R. R. In: Oxygen Free Radicals and Tissue Damage, Ciba. Fdn. Symposium 65 (New series): Amsterdam; Excerpta Medica, p. 57 (1979).
113. Blake, D. R., Hall, N. D., Bacon, P. A., Halliwell, 8., and Gutteridge, V. M. c., Lancet ii:1I42 (1981).
114. Svoboda, P., and Mosinger, B., Biochem. Pharm.30:2243 (1981).
15. Halliwen:B., FEBS Letters 92:321 (1978).
116. Carrell, R. W., Krishnamoorthy, R. and Winterbourn, C. C. In: The Red Cell. Brewer, G. J., ed., A. R. Liss, New York, p. 687 (1978).
117. Brunori, M., Fa1cioni, G., Fioretti, E., Gardina, 8., and Ritilio, G., Eur. J. Biochem. 53:99 (1975).
118. Delmelle, M., Photochem. Photobiol. 27:731 (1978).
119. Anderson, S. M. and Krinsky, N. I., Photochem.Photobiol. 18:403 (1973).
120. Allen, R. E., Stjernholm, R. L., Steele, R. H., Biochem. Biophys. Res. Commun. 47:679(1972).
121. Cilento, G., J. Theor. BioI. 55,471 (1975).
122. Koppenol, W. H., Nature 262:410 (1976).
123. Flohe, L. In: Oxygen Free Radicals and Tissue Damage, Ciba, Symp. 65 (New Series), Amsterdam, Exerpta Medica, p. 95 (1979).
124. Kosower, N. S., and Kosower, E. M. In: Free Radicals in Biology, ed. W. A. Pryor, Academic Press, New York, p. 55 (1976).
125. Fridovich, I. In: Oxygen Free Radicals and Tissue Damage, Ciba Symp, 65 (New series) 77 (1979).
126. Halliwell, B., New Phytologist 73:1075 (1974).
127. Jakoby, W. 0. and Keen, J. H., Trends in Biological Sciences 2:229 ( 1977).
128. Dendo, C. W., Agents Actions 9:333 (1979).
129. Michelson, A. F., and Puget, K. Acta Physiol Scand suppl. 492:67 (1980).
130. Ames, 0. N., Catheart, R., Schwiers, E., and Hochstein, P., Proc. Natl. A cad. Sci. USA, 78:6858 (1981).
131. Bourgain, R. H., Deby, c., Deby-Dupont, G., and Andries, R., Biochem. Pharm. 31:3011 (1982).
132. Matsushita, S., Ibuka, F. and Aoki, A., Arch.Biochem. Biophys. 102:446 (1963).
133. Proctor, P. H., Nature, 228:868 (1970).
134. Pullman, B., and Pullman, A. Quantum Biochemistry, Academic Press, New York, p. 217 (1963).
13S. Brabham, D. E., and Lee, J., J. Phys. Chem.
80:2292 (1976).
136. Montgomery, M. R., Furry, J., Gre, S. J., and Krieger, R. I., Toxicity and Applied Pharmacology 63:32 (1982).
137. Proctor, P., Lancet, ii:95 (1978).
138. Lowrey, J. C., Veterinary Medicinef Small Animal Clinicians, 71:289 (1976).
139. Hilton, J. G., Brown, G. L., and Proctor, P. H., Toxicology and Applied Pharmacology. 53:50 (1980).
140. Bucher, J. R. and Roberts, R. J., Pediatric Pharmacology 2:1 (1982).
141. Gray, B., Tox. and Appl. Pharm. 60:479 (1981).
142. Katzenstein, A. L., Bloor, C. M., Leibow, A. A., Am. J. Path. 85:210 (1976).
143. Mais, D. E., Lahr, P. D., Bosin, T. R., Toxicology and Applied Pharmacology 64:221 (1982).
144. Frank, L., Yam, J., and Roberts, R. J., J. Clin.Invest. 61:269 (1978).
145. Gray, B., Tox. Appl. Pharm. 60:479 (1981).
146. Rinaldo, J. E. and Rogers, R. M., N. Engl. J.Med. 306:900 (1982).
147. McLennon, G. and Autor, R. P. In: Pathology of Oxygen. Autor, A. P., ed., New York, Academic Press, p. 85 (1982).
148. Edsmyr, F., Huber, W., and Menander, K. B., Curro Ther. Res. 19:198 (1976).
149. Phillips, T. L., Seminars in Oncology 8:65 (1981).
150. Halliwell, B., Trends in Biochemical Sciences 7:270 (1982).
151. Fee, J. A., Trends in Biochemical Sciences. 7:84 (1982).
152. Sacks, T., Moldow, C. F., Craddock, P. R., Bowers, T. K., and Jacob, H. S., Prog. Clin. Bioi.Res. 21:719 (1978).
153. Pottathil, R., Chandrabose, K. A., Cuatrecasas, P., and Lang, D. J., Proc. Natl. Acad. Sci. USA.78:3343 (1981).154. McCord, J. M., Wong, K., Stokes, S. H., Petrone, W. F., English, D. In: Pathology of Oxygen, Autor, A. P., ed., New York, Academic Press, p.
75 (1982).55. McCord, J. M., and English, D. In: Enzymes as Drugs, ed. U. S. Hollenberg and V. Roberts, New York, Wiley Interscience, p. 353 (1981).
156. Clark, R. A., Szot, S., Venkatasubramanian, K., Schiffman, E., J. Immunol.124:2020 (1980).
157. Emerit, I., and Ceruti, P., Proc. Natl. A cad. Sci.USA. 78:1868 (1981).
158. Emerit, I., and Michelson, A. M., Proc. Natl.Acad. Sci. USA 78:2537 (1981).
159. Emerit, I., and Michelson, A. M., Physiol. Scand.suppl 492:59 (1980).
160. Aune, T. M., and Pierce, C. W., Proc. Nat/. Acad.Sci. USA. 78:5099 (1981).
161. Carp, H., and Jannoff, A., J. Clin. Invest. 63:793 (1979).
162. Campbell, E. J., Senior, R. M., McDonald, J. A., and Cox, D. L., J. Clin. Invest. 70:845 (1982).
163. Matheson, N. R., Wong, P. S., Schuyler, M., and Travis, J., Biochemistry 20:331 (1981).
164. Clark, R. A., Stone, P. J., Hair, A. E., Calore, J.D., J. BioI. Chem. 256:3348 (1981).
165. Oyanagui, Y., Biochemical Pharmacology, 30:1791 (1982).
166. Del Maestro, R. F., Bjork, J. and Arfors, K. E. In: Pathology of Oxygen. Autor, A. P., ed., Aca-demic Press, New York, p. 157 (1982).
167. Clark, R. A. and Klebanoff, S. J., J. Immunol.124:399 (1980).
168. Handin, R. I., Karabin, R., and Boxer, G., J.Clin. Invest. 59:959 (1977).
169. In: Pathology of Oxygen, Autor, A. P., ed., New York Academic Press, p. 85 (1982).
170. White, J. G., Ruq, G. H. R., and Gerrard, J. M., Am. J. Path. 88:387 (1977).
171. EI-Sabban, F., and Rossenblum, W. I., Stroke, 13:35 (1982).
172. Huyssain, M. Z., and Bhatmagar, R. S., Biochem.Biophys. Res. Commun. 89:71 (1979).
173. Pick, E., Keisari, Y., Jakebowski, A., Bromberg, Y., and Freund, M., Lymphokines and Thymic Hormones: Their Potential Utilization in Cancer Therapeutics, eds., A. L. Goldstein and M. A.Chirigos, Raven Press, New York, p. 177 (1981).
174. Nishida, Y., Tanimoto, K., and Akaoka, I., Clinicallmmunology and Immunopathology 19:319 (1981).
175. Ohmori, H., Komoriya, K., Azuma, A., Korozumi, S., Yoshinobu, H. 0., Biochem. Pharm., 28:333 (1979).
176. Bragt, P. C and Bonta, I. L., Agents and Actions, 10:536 (1980).
177. Hirschelmann, R., and Bekemeier, H., Experientia, 37:1313 (1981).
178. Puig-Parellada, P., and Planas, J. M., Biochem.Pharm. 25:535 (1976).
179. White, G. J., Agents and Actions, 11:503 (1981).
180. Pekoe, G., VanDyke, K., Peden, D., Mengoli, H., and English, D., Agents and Actions. 12:371 (1982).
181. Weissman, G., J. Lab. C/in. Med. 100:322 (1982).
182. Jimenez, A. H., and WiIlkens, R. F., J. Lab. Clin.Med. 100:489 (1982).
183. Berton, G., Schneider, C, and Romes, D., Biochim. Biophys. Acta. 595:47 (1980).
184. Huber, W. and Schulte, T. L., Pharmaceutical Compositions Comprising Orgotein and their use. U. S. Pat. No. 3,773,928 (1973).
185. Huber, W., Menander-Huber, K. B., Saifer, M. G. P., and Dang, P. H-C In: Perspectives in Inflammation, eds. D. A. Willoughby, J. P. Giraud, and J. P. Velo, Baltimore, University Park Press, p. 527 (1977).
186. Petkau, A., Checlack, W. S., Kelley, K., Friesen, H. G. In: Pathology of Oxygen, Autor, A. P., ed., Academic Press, New York, p. 223 (1982).
187. Villasor, R. P. In: Pathology of Oxygen, Autor, A. P., ed., Academic Press, New York, p. 303 (1982).
188. Edsmyr, F. In: Pathology of Oxygen, Autor, A.P., ed., Academic Press, New York, p. 315 (1982).
189. Bartsch, G., and Marberger, H. In: Pathology of Oxygen, Autor, A. P., Academic Press, New York, p. 327 (1982).
190. Lund-Oleson, K. In: Pathology of Oxygen, Autor, A. P., Academic Press, New York, p. 339 (1982).
191. Schmidt, J. D., and Schulte, T. L. In: Pathology of Oxygen, Autor, A. P., Academic Press, New York, p. 355 (1982).
192. Oyanagui, Y., Biochem. Pharmacol. 25:1465 (1976).
193. Lund-Olesen, K., and Menader, K. B., Curro Therap. Res. 16:706 (1974).
194. Huber, W., Schulte, T. L., Carson, S., Goldhamer, R. E. and Yogin, E. E., Toxicol. Appl.Pharmacol. 12:308 (1968).
195. Riu, R., LeDen, R., and LeMoriel, C, Med. Ind.6:811 (1971).
196. Fuenfer, M. M., Carr, E. A., and Polk, H. C, J.Surg. Res. 27:29 (1979).
197. Greenwald, R. A., Journal of Rheumatology, Suppl. 1l:9 (1981).
198. Sorenson, J. R., J. Med. Chem. 19:135 (1976).
199. Oyanagui, Y., Biochem. Pharm., 27:777 (1978).
200. Oyanagui, Y., Biochem. Pharmacol. 25:1473 (1976).
201. Takenaka, T., Okuda, M., Kawabori, S., Kubo, K., Clin. Exp. Immunol. 28:56 (1977).
202. Gampp, H. and Zuberbuhler, A. D. In: Metal Ions in Biological Systems, Vol. 12, ed: Helmut Siegel, Marcel Decker, New York, p. 133 (1981).
203. Abramson, S., Hoffstein, S. T., and Weissman, G., Arthritis and Rheumatism 25:174 (1982).
204. Simchowitz, L., Atkinson, J. P., and Spilberg, I., Arthritis and Rheumatism, 25:181 (1982).
205. Jacob, J. S., Craddock, P. R., Hammerschmidt, D. E., and Moldow, C. F., N. Eng. J. Med., 302:789 (1980).
206. Chan, P. H., and Fishman, R. A., J. Neurochem.35:1004 (1980).
207. Bahri, A. K., Chiang, C. S., and Timbrell, J. A., Taxicol. Applied. Pharmacol. 60:561 (1981).
208. Winston, G. W., and Cedarbaum, A. I., Biochem.Pharm. 31:2031 (1982).
209. Walker, E. M., Gale, G. R., Ann. Clin. Lab. Sci., 1l:397 (1981).
210. McGinness, J. E., Grossie, B. V., Benjamin, R. S., Hokanson, J. A., in submission.
211. Westman, N. G. and Marklund, S. L., Cancer Research 41:2962 (1981).
212. Tritton, T. R. and Yee, G., Science, 217:248 (1982).
213. Tritton, T. R., Yee, G., Windard, L. B., Fedn.Proc. 42:284 (1983).
214. Daskal, Y., Woodard, C, Crooke, S. T., and Busch, H., Cancer Res. 38:467 (1978).
21S. Oberley, L. W., Leuthauser, S., Beutner, G. R., Sorenson, J. R. J., Oberley, T. D., Bize, I. B., In:
Pathology of Oxygen, Autor, A. P., ed., Academic Press, New York, p. 207 (1982).
216. Michaelson, A., Personal communication, 217. Till, G. 0., Johnson, K. J., Kunkel, R., and Ward, P. A., J. Clin. Invest. 69:1126 (1982).
218. Suttop, N., and Simon, L. M., J. Clin. Invest.70:3~2 (1982).
219. Rebello, G. and Mason, J. K., Histopathology 1:53 (1978).
220. Rosenthal, A. R., Appleton, B., and Hopkins, J.L., Am. J. Ophthal. 78:671 (1974).
221. Autor, A. P., Life Sci. 14:1309 (1974).
222. Matkovics, B., Barabas, K., Varga, Sz. I., Szabo, L. and Berencsi, G., Gen. Pharmac. 13:333 ( 1982).
223. Cotzias, G. C., Papavasiliou, P. S., Gellene, R. Aronson, R. B., and Mena, I. In: 3rd Symposium on Parkinson s Disease, London, E. and S. Livingston Ltd., 178 (1969).
224. Bridelli, M., Capelletti, R., and Crippa, P. R., Bioectrochemistry and Bioenergetics 7:555 (1981).
225. Mitzutani, D., Massalski, T. B., McGinness, J. E., Corry, P., Nature, 259:505 (1976).
226. Cotzias, G. C., Developments in Treatments for Parkinson s Disease, New York, Medcom Press, p. I (1971).
227. Altshule, A., Clinical Pharmacology and Therapeutics, 19:124 (1975).
228. LaFerriere, K. A., Arenberg, I. K., Hawkins, J.
E., Johnson, L-G., Ann. 0101.83:685 (1974).
229. Larsson, B., Acta Universitatis Upsaliensis, 43:1 (1979).
~O. Lindquist, N. G., Acta. Radiol. Scand. suppl.325:1 (1973).
231. Broun, K. S., Bergsma, D. K., Barrow, M. V., In:Ear. Birth Defects: original article series, Vol.VII, (4) (1971).
232. Kono, R., Yamaoka, T., Yoshizaki, H., and McGinnis, J., J. Appl. Phys. 50:1236 (1979).
233. Pullman, A., and Pullman, B., Biochim. Biophys.Acta. 54:384 (1961).
234. McGinness, J. E., Corry, P. M., and Proctor, P.H., Science, 183:853 (1974).
235. Wasserman, H. P., Ethnic Pigmentation, Exerpta Medica, Amsterdam, and American Elsevier, p.244 (1974).
236. Cotzias, G. C., Developments in Treatments for Parkinson s Disease, New York, Medcom Press,
237. Creel, D., Pharmacology, Biochemistry, and Behavior, 12:969 (1980).
238. Barden, H. In: Aging, Vol. I, eds: Brody, H., Harman, D., and Ordy, J. M., Raven Press, New York, p. 79 (1975).
239. Proctor, P., Lancet. i:1069 (1971).
240. Kastin, A. J., Kuzemchak, B., Tompkins, R. G., Schally, A. V., and Miller, C. M., Brain Res.
Bull. 1:567 (1976).
241. Lacey, M. E., Physiol. Chem. Phys. 13:319 (1981).
242. Hood, V. D., Poole, J. P., Freedman, L., Audiology, 15:449 (1981).
243. Darwin, C. The Variation of Animals and Plants Under Domestication, Vol. 2. New York, D. Appleton, p. 322 (1896).
244. Donaldson, J., Labella, F. S., and Gessor, D., Neurotoxicology 2:53 (1980).
245. Mann, D. M. A., and Yates, P. D., Arch. Neurol.39:545 (1982).
246. Cohen, G. In: Pathology of Oxygen, Autor, A. P., ed., Academic Press, New York, p. 115 (1982).
247. Tse, D. C. S., McGreery, R. L., and Adams, R.N., Journal of Medicinal Chemistry, 19:37 (1976).
248. Tiffany-Castiglione, E., Saneto, R. P., Proctor, P.H., and Perez-Polo, J. R., Biochem. Pharm. 31:181 (1982).
249. Yoffe, J. R., and Borchandt, R. T., Life Sciences.31:489 (1982).
250. Cohen, G., and Heikkila, R. E., J. BioI. Chem. 249:2447 (1974).
251. Bristow, M. R., Minobe, W. A., Billingham, M.E., Marmon, J. B., Sageman, W. S., and Daniels, J. R., Lab. Invest. 45:157 (1981).
252: Perrot, H., and Ortonne, J. P., Arch. Derm. Res.261 :245 (1978).
253. Pratt, C. B., and Shanks, E. C., J. Amer. Med.Ass. 228:460 (1974).
254. Boekelheide, K., Graham, D. G., Mize, P. D., and Vogel, F. S., Am. J. Path. 100:651 (1980).
255. Kelman, S. N., Sullivan, S. G., and Stern, A., Biochem. Pharm. 31:2409 (1982).
256. Aebi, H. and Suter, H., Acatalasemia in Stanbury, J. B., Wyngaarden, J. B., and Fredrickson, D. S. The Metabolic Basis of Inherited Disease.
McGraw-Hili, New York, p. 1710 (1972).
157. Rister, M., Bauermeister, K., Gravert, D., Gladtke, E., Lancet. i:1094 (1978).
258. Howes, R. M., Allen, R. C., Su, C. T., Hoopes, J.
E., Surgical Forum, 27:558 (1976).
259. Pauling, Linus. Trends in Biochemical Sciences, 4. (1979).
260. Mann, T. and Keilin, D., Proc. R. Soc. London, Ser. B., 126:303 (1939).
261. Commoner, B., Townsend, J. and Pake, G. E., Nature. 174:689 (1952).
262. McCord, J. M. and Fridovich, I., J. Bioi. Chem.
224:6049 (1969).
263. Marklund, S. L., J. Bioi. Chem. 251:7504 (1976).
264. Nishikimi, M., Appaji, N., and Yagi, K., Biochem. Biophys. Res. Commun. 46:849 (1972).
265. Ringel, S. P., Stern, L. Z., Menander, K. 8., lonaesai, Y., Lava, N. S., Pellegrino, R. J., Snyder, R. D., Ziter, F. A., Allsop, K. G., In: Muscular Dystrophy Research: Adavnces and New Trends, eds: C. Angelini, G. A. Daniels, and D. Fontanari, Excepta Medica, Princeton, p. 198 (1980).
(Received June 25, 1984).
*Department of Pathology, University of Texas Medical School, Houston, Houston, Texas 77030.
**Laboratory of Chemical Pathology, Department of Pathology, University of Texas Medical Branch, Galveston, Texas (Deceased).
My coauthor on this review, Edward Reynolds, died suddenly in November of 1983. Dr. Reynolds was chairman of the Department of Pathology at the University of Texas Medical Branch in Galveston. He was also a major figure in the area of the free radical-mediated toxicity of drugs, having done much of the early work on carbon tetrachloride and halothane. Significantly, shortly before his death, he was branching out to the area of NMR, looking for new worlds to explore. Lt is thus fitting that this, one of his final works, should be published in the journal that published much of the original work on NMR.
Supported in part by a grant AM-27135 from the National Institutes of Health and by Grants from the Retina Research Foundation, Houston, and the Alexander Medical Foundation, San Carlos, California.
*** Flamm ES, Demopoulos HB, Seligman ML, Poser RG, Ransohoff J.,“Free radicals in cerebral ischemia” Stroke. 1978 Sep-Oct;9(5):445-7.
“The possibility that cerebral ischemia may initiate a series of pathological free radical reactions within the membrane components of the CNS was investigated in the cat. The normally occurring electron transport radicals require adequate molecular oxygen for orderly transport of electrons and protons. A decrease in tissue oxygen removes the controls over the electron transport radicals, and allows them to initiate pathologic radical reactions among cell membranes such as mitochondria. Pathologic radical reactions result in multiple products, each of which may be present in too small a concentration to permit their detection at early time periods. It is possible to follow the time course, however, by the decrease of a major antioxidant as it is consumedby the pathologic radical reactions. For this reason, ascorbic acid was measured in ischemic and control brain following middle cerebral artery occlusion. There was a progressive decrease in the amount of detectable ascorbic acid ranging from 25% at 1 hour to 65% at 24 hours after occlusion. The reduction of this normally occurring antioxidant and free radical scavenger may indicate consumption of ascorbic acid in an attempt to quench pathologic free radical reactions occurring within the components of cytomembranes.”
Keywords: